

New enzymes and their role in disease
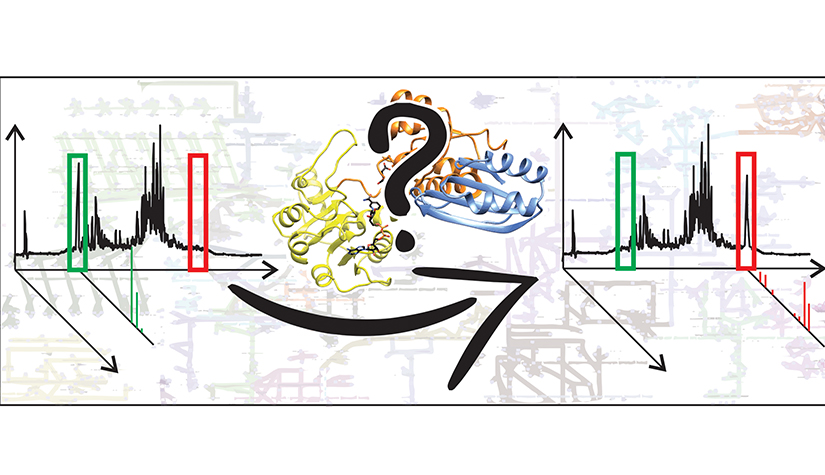
The Enzymology & Metabolism group, led by Associate Prof. Carole Linster, deciphers the cellular metabolism in rare and more common neurodegenerative diseases.
A glimpse into our research

Filling gaps in metabolic networks to understand diseases
Our understanding of cellular metabolism and its regulation is far from being complete. We exploit genomic and post-genomic data to fill gaps in metabolic networks, discover new enzyme functions, and elucidate their role in disease. Our group focuses on rare neurometabolic diseases, but occasionally expands to more common diseases, depending on where our enzymes and proteins of interest lead us.