

Deciphering molecular underpinnings of Parkinson’s disease
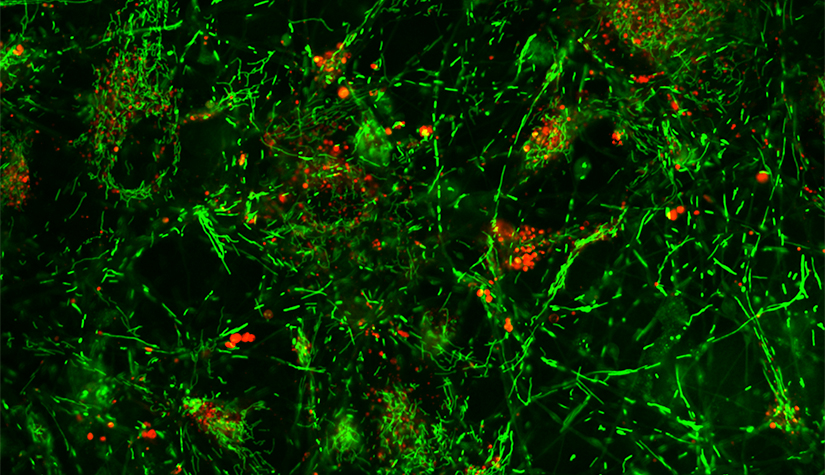
The Molecular & Functional Neurobiology group, led by Prof. Anne Grünewald, studies genetic and non-genetic causes of neurodegeneration in Parkinson’s disease.
A glimpse into our research

We explore the role of the mitochondrial genome in idiopathic Parkinson’s disease. Our aim is to identify cellular pathways, which may be targeted for therapeutic intervention. For monogenic Parkinsonism, we aim to understand why some individuals do not manifest any signs of the disease despite the presence of a mutation. We want to uncover mechanisms that could be modulated to delay the onset.
News
-

Federal President Steinmeier and HRH Grand Duke Henry visit the LCSB
OutreachLife Sciences & MedicineLearn more